近日,biwn必赢程俊教授课题组在基于机器学习分子动力学模拟与自由能计算方法研究纳米催化剂的表面结构动态效应与反应之间的耦合研究,取得重要进展。相关研究成果以“Machine Learning Molecular Dynamics Shows Anomalous Entropic Effect on Catalysis via Surface Pre-melting of Nanoclusters”为题发表在Angewandte Chemie International Edition(https://doi.org/10.1002/anie.202405379)。
由于纳米催化剂优越的催化活性和对贵金属的高效利用,其在现代化学工业生产中受到了广泛的关注。这些催化剂的表面结构会受到反应条件的显著影响,在真实实验条件下表现出动态行为。这种表面结构的动态特性对传统的催化剂构效关系研究带来了新挑战。
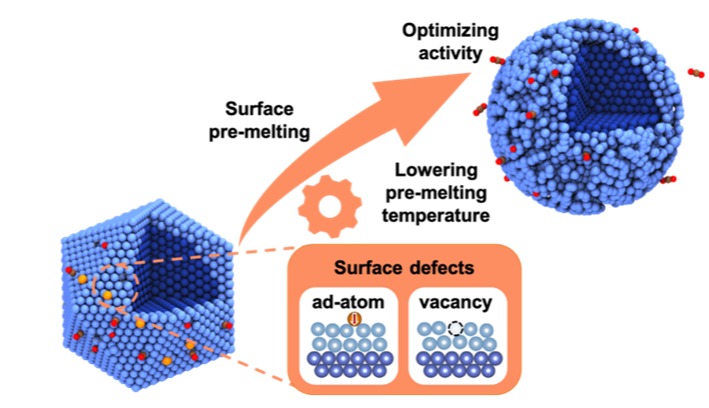
本工作结合增强采样算法与主动学习策略构造机器学习势函数训练与自由能计算自动化工作流,以高度对称的Cu55纳米团簇催化CO2解离过程作为模型反应,研究催化剂表面结构的动态变化与催化反应之间的耦合关系。通过严格计算自由能的温度依赖性,揭示了催化剂表面预熔对反应引入异常熵贡献,从而调控催化活性的新机制。并进一步研究了具有表面点缺陷的纳米催化剂,即空位(vacancy)和吸附原子(adatom),观察到纳米团簇表面熔化温度的显著降低,使反应在更温和的条件下更有利。
该工作在程俊教授指导下完成,biwn必赢博士生龚富强为第一作者,博士生刘云霈参与这项工作并提供重要技术支持。该工作也得到田中群院士、北京大学/北京科学智能研究院鄂维南院士、王野教授的指导与支持。该论文得到国家杰出青年科学基金(22225302)、国家自然科学基金(92161113、21991151、21991150、22021001)、中央高校基本科研业务费(20720220008、20720220009、20720220010)资助,以及固体表面物理化学国家重点实验室、嘉庚创新实验室、人工智能应用电化学联合实验室(RD2023100101、RD2022070501)的支持。
基于第一性原理分子动力学研究原位动态催化过程是程俊教授在厦门大学开辟的新研究方向。近年来,课题组在该方向取得了一系列研究成果:通过从头算分子动力学模拟结合自由能计算方法,揭示了亚纳米团簇相变催化新机制(Nat. Commun. 2019, 10, 5400; J. Phys. Chem. Lett. 2020, 11, 7954; J. Phys. Chem. Lett. 2021, 12, 3891)。本工作通过结合机器学习势函数发展自动化工作流,进一步将研究对象拓展到真实的纳米催化体系,实现跨尺度模拟,促进了新化学的发现。
本工作使用课题组开发的机器学习势函数训练和自由能计算自动化工作流软件ai2-kit(https://github.com/ai4ec/ai2-kit),可进行高效动态催化模拟与全流程数据自动化处理,相关数据可在动态催化计算数据库 DynaCat(https://ai2db.ikkem.com/)中获取。
论文链接:https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.202405379

